Welcome to Alex and Emily who will be joining us for their projects for the next couple of months.

Welcome to Alex and Emily who will be joining us for their projects for the next couple of months.
Welcome to Alex who has just started his Phd in our group.

Welcome to Pavi who has started as a postdoc in our group.

A large segment of the proteome consists of disordered regions, yet in most cases, little is known about their mechanisms and functions. What are the roles of protein disorder in cell biology, and how do intrinsically disordered proteins function? These are the questions Cell’s Robert Kruger posed to Madan Babu, Julie Forman-Kay, and Richard Kriwacki. Annotated excerpts from this conversation are presented below, and the full conversation is available with the article online.
Gene fusions are common cancer-causing mutations, but the molecular principles by which fusion protein products affect interaction networks and cause disease are not well understood. Here, we perform an integrative analysis of the structural, interactomic, and regulatory properties of thousands of putative fusion proteins. We demonstrate that genes that form fusions (i.e., parent genes) tend to be highly connected hub genes, whose protein products are enriched in structured and disordered interaction-mediating features. Fusion often results in the loss of these parental features and the depletion of regulatory sites such as post-translational modifications. Fusion products disproportionately connect proteins that did not previously interact in the protein interaction network. In this manner, fusion products can escape cellular regulation and constitutively rewire protein interaction networks. We suggest that the deregulation of central, interaction-prone proteins may represent a widespread mechanism by which fusion proteins alter the topology of cellular signaling pathways and promote cancer. Paper by Natasha Latysheva et al can be found here.

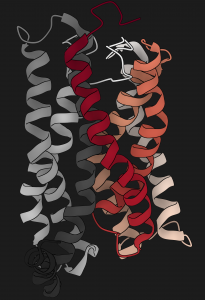
Class A G-protein-coupled receptors (GPCRs) are a large family of membrane proteins that mediate a wide variety of physiological functions, including vision, neurotransmission and immune responses. They are the targets of nearly one-third of all prescribed medicinal drugs such as beta blockers and antipsychotics. GPCR activation is facilitated by extracellular ligands and leads to the recruitment of intracellular G proteins. Structural rearrangements of residue contacts in the transmembrane domain serve as ‘activation pathways’ that connect the ligand-binding pocket to the G-protein-coupling region within the receptor. In order to investigate the similarities in activation pathways across class A GPCRs, we analysed 27 GPCRs from diverse subgroups for which structures of active, inactive or both states were available. Here we show that, despite the diversity in activation pathways between receptors, the pathways converge near the G-protein-coupling region. This convergence is mediated by a highly conserved structural rearrangement of residue contacts between transmembrane helices 3, 6 and 7 that releases G-protein-contacting residues. The convergence of activation pathways may explain how the activation steps initiated by diverse ligands enable GPCRs to bind a common repertoire of G proteins. Paper by AJ Venkatakrishnan et al can be found here.

The proteasome has pronounced preferences for the amino acid sequence of its substrates at the site where it initiates degradation. Here, we report that modulating these sequences can tune the steady-state abundance of proteins over 2 orders of magnitude in cells. This is the same dynamic range as seen for inducing ubiquitination through a classic N-end rule degron. The stability and abundance of His3 constructs dictated by the initiation site affect survival of yeast cells and show that variation in proteasomal initiation can affect fitness. The proteasome’s sequence preferences are linked directly to the affinity of the initiation sites to their receptor on the proteasome and are conserved between Saccharomyces cerevisiae, Schizosaccharomyces pombe, and human cells. These findings establish that the sequence composition of unstructured initiation sites influences protein abundance in vivo in an evolutionarily conserved manner and can affect phenotype and fitness. Paper by Yu H et al can be found here.


EMBO elects new members annually on the basis of scientific excellence and outstanding research contributions. The organisation promotes excellence in life sciences by supporting talented researchers, and stimulating exchange of scientific information. Madan, along with 57 other researchers, joins more than 1700 of the best researchers from Europe and around the world. Please see here for the EMBO and here for the LMB press release.
Although gene fusions have been recognized as important drivers of cancer for decades, our understanding of the prevalence and function of gene fusions has been revolutionized by the rise of next-generation sequencing, advances in bioinformatics theory and an increasing capacity for large-scale computational biology. The computational work on gene fusions has been vastly diverse, and the present state of the literature is fragmented. It will be fruitful to merge three camps of gene fusion bioinformatics that appear to rarely cross over: (i) data-intensive computational work characterizing the molecular biology of gene fusions; (ii) development research on fusion detection tools, candidate fusion prioritization algorithms and dedicated fusion databases and (iii) clinical research that seeks to either therapeutically target fusion transcripts and proteins or leverages advances in detection tools to perform large-scale surveys of gene fusion landscapes in specific cancer types. In this review, we unify these different-yet highly complementary and symbiotic-approaches with the view that increased synergy will catalyze advancements in gene fusion identification, characterization and significance evaluation. Paper by Natasha Latysheva and M. Madan Babu can be found here.

Dr Mark Bayfield is a sabbatical visitor from Dept of Biology, York University, Toronto and has joined our group for 5 months to learn techniques in computational biology (data integration and analysis of next generation sequencing data).
