Welcome to Andal who has just started as a postdoc in our group.

Welcome to Andal who has just started as a postdoc in our group.
Cell-to-cell variation in gene expression levels (noise) generates phenotypic diversity and is an important phenomenon in evolution, development and disease. TATA-box binding protein (TBP) is an essential factor that is required at virtually every eukaryotic promoter to initiate transcription. While the presence of a TATA-box motif in the promoter has been strongly linked with noise, the molecular mechanism driving this relationship is less well understood. Through an integrated analysis of multiple large-scale data sets, computer simulation and experimental validation in yeast, we provide molecular insights into how noise arises as an emergent property of variable binding affinity of TBP for different promoter sequences, competition between interaction partners to bind the same surface on TBP (to either promote or disrupt transcription initiation) and variable residence times of TBP complexes at a promoter. These determinants may be fine-tuned under different conditions and during evolution to modulate eukaryotic gene expression noise. Paper by Charles Ravarani et al can be found here.

Congratulations to our postdoc Rita to be elected as a Fellow of Darwin College!
Big congratulations to Tilman to win this year’s Max Perutz Student Prize.
The Max Perutz Student Prize is awarded annually for outstanding work performed at the LMB prior to the award of a PhD. The 2015 prize has been awarded to Tilman Flock, for his comprehensive computational analysis of GTP-binding proteins (G proteins), revealing the universal nature of their interactions and activation.
Tilman, a third year PhD student in the group of Madan Babu in the Structural Studies Division, undertook a systematic analysis of over 80 structures and 950 sequences of G proteins from different species to reveal how the core mechanism of activation and recognition is conserved, even while new specific interactions evolve. In humans there are over 800 G protein-coupled receptors which, upon binding of an extracellular ligand, activate one or more of 16 different G proteins by triggering the exchange of GDP for GTP, thus initiating a series of signalling pathways. It is these receptors that allow us to smell different chemicals, respond to adrenalin, and sense neurotransmitters in the brain, amongst many other functions. More than 30% of all prescribed small molecule drugs act by stimulating or inhibiting them. Since the 1980’s, there have been over 11,000 publications relating to how G proteins work. Tilman’s elegant methodological approach unifies a vast amount of data, providing a framework for the whole field. It is a general approach that can be extended to other proteins, and it shows the power of large-scale analysis of the ever-growing mass of published data.
See Flock et al, Nature 524, 173-179 (2015).
The research student prize is awarded by the Max Perutz Fund. The Fund was established for the promotion and advancement of education and research in molecular biology and allied biomedical sciences.

Photo and text are taken from the LMB website.
COSB issue is now published! You can find the article by Anne R Panchenko and M. Madan Babu here.

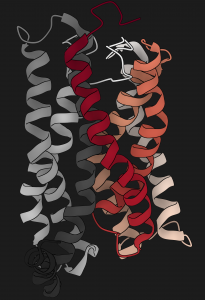
We present comprehensive maps at single-amino acid resolution of the residues stabilizing the human Gαi1 subunit in nucleotide- and receptor-bound states. We generated these maps by measuring the effects of alanine mutations on the stability of Gαi1 and the rhodopsin-Gαi1 complex. We identified stabilization clusters in the GTPase and helical domains responsible for structural integrity and the conformational changes associated with activation. In activation cluster I, helices α1 and α5 pack against strands β1-β3 to stabilize the nucleotide-bound states. In the receptor-bound state, these interactions are replaced by interactions between α5 and strands β4-β6. Key residues in this cluster are Y320, which is crucial for the stabilization of the receptor-bound state, and F336, which stabilizes nucleotide-bound states. Destabilization of helix α1, caused by rearrangement of this activation cluster, leads to the weakening of the interdomain interface and release of GDP. The paper by Dawei Sun, Tilman Flock et al can be viewed here.

Excessive expansions of glutamine (Q)-rich repeats in various human proteins are known to result in severe neurodegenerative disorders such as Huntington’s disease and several ataxias. However, the physiological role of these repeats and the consequences of more moderate repeat variation remain unknown. Here, we demonstrate that Q-rich domains are highly enriched in eukaryotic transcription factors where they act as functional modulators. Incremental changes in the number of repeats in the yeast transcriptional regulator Ssn6 (Cyc8) result in systematic, repeat-length-dependent variation in expression of target genes that result in direct phenotypic changes. The function of Ssn6 increases with its repeat number until a certain threshold where further expansion leads to aggregation. Quantitative proteomic analysis reveals that the Ssn6 repeats affect its solubility and interactions with Tup1 and other regulators. Thus, Q-rich repeats are dynamic functional domains that modulate a regulator’s innate function, with the inherent risk of pathogenic repeat expansions. The paper by Rita Gemayel, Sreenivas Chavali et al can be viewed here.

Many congratulations to Madan to be awarded the prestigious the Royal Society Francis Crick Medal and Lecture 2016. Madan will deliver the prize lecture in December 2016. Here is the detailed information as shown in the LMB website. You can get more information about the Royal Society Awards Francis Crick Medal here.
G protein-coupled receptors (GPCRs) allosterically activate heterotrimeric G proteins and trigger GDP release. Given that there are 800 human GPCRs and 16 different Ga genes, this raises the question of whether a universal allosteric mechanism governs Ga activation. Here we show that different GPCRs interact with and activate Ga proteins through a highly conserved mechanism. Comparison of Ga with the small G protein Ras reveals how the evolution of short segments that undergo disorder-to-order transitions can decouple regions important for allosteric activation from receptor binding specificity. This might explain how the GPCR–Ga system diversified rapidly, while conserving the allosteric activation mechanism. The paper by Tilman Flock et al can be viewed here.

The proteasome controls the concentrations of most proteins in eukaryotic cells. It recognizes its protein substrates through ubiquitin tags and initiates degradation at disordered regions within the substrate. Here we show that the proteasome has pronounced preferences for the amino acid sequence of the regions at which it initiates degradation. Specifically, proteins in which the initiation regions have biased amino acid compositions show longer half-lives in yeast than proteins with unbiased sequences in the regions. The relationship is also observed on a genomic scale in mouse cells. These preferences affect the degradation rates of proteins in vitro, can explain the unexpected stability of natural proteins in yeast and may affect the accumulation of toxic proteins in disease. We propose that the proteasome’s sequence preferences provide a second component to the degradation code and may fine-tune protein half-life in cells.The paper by Susan Fishbain, Sreenivas Chavali et al can be viewed here.
